TL;DR: A COA (Certificate of Analysis) is a laboratory report assigned to a specific batch/lot that documents testing methods, acceptance criteria, and results. Reading a COA correctly involves verifying batch traceability, understanding the method (e.g. HPLC, MS), interpreting units and limits of detection, and being aware of the report’s limitations.
What Is a COA (Certificate of Analysis)?
COA (also written as CoA) stands for Certificate of Analysis — a formal laboratory report pertaining to a specific batch/lot of material. The document presents the results of analytical testing along with key identifying information (e.g. batch number, sample identifier, dates), enabling an assessment of conformance against defined requirements or specifications.
A COA is not a general description of the material or a declaration of „universal quality.” Its informational value depends on who performed the tests, how the sample was collected and controlled, what methods were used, and what criteria were applied to evaluate the results.

Why Correct Interpretation of a COA Matters in Quality and Compliance
Reading and understanding a COA correctly is important because it affects:
- Traceability: whether the results actually refer to the batch you hold.
- Quality decisions: whether the batch meets defined criteria (rather than intuitive expectations).
- Documentary consistency: whether the document is useful in an audit and quality system (methods, report numbers, authorisation).
- Risk management: whether you understand the limitations (scope of analytes, sample representativeness, measurement uncertainty).
From a compliance perspective, a COA is evidence „within the scope it describes” — it supports conformance assessment only when it is consistent, traceable, and methodologically transparent.
How to Read a COA? Certificate of Analysis Step by Step
The practical order of COA review is: identification and traceability → scope and methods → specification → results → data context → document authorisation.
1) Verifying Traceability (batch number on certificate of analysis)
Check that the COA contains at least:
- the material name and any internal code or designation,
- the batch/lot number,
- the sample identifier (sample ID),
- the date(s) of testing and/or sampling,
- the issuing entity (laboratory / manufacturer) and report number.
What to confirm
- The batch number on the COA must match the batch number on the container label and accompanying documents.
- Dates should be logically consistent (e.g. the testing date should not „precede” the manufacturing date without an explanation such as a retest).
- If multiple parties are involved in the chain (manufacturer, packager, third-party laboratory), the COA should clearly indicate each party’s role.
2) Who Issued the COA: Manufacturer or Third-Party Laboratory?
A COA may originate from:
- the manufacturer’s in-house QC department,
- an external contract (third-party) laboratory,
- an intermediary entity that re-issues results (reissued COA).
Why this matters
Differences in sampling, chain of custody, and document control all affect how the credibility and scope of conclusions should be interpreted. The document should indicate whether the results are original or cited from another report (including the source report number).
3) Specification and Acceptance Criteria: „Conforms” Is Not Enough Without Context
A well-prepared COA shows:
- the test name,
- the criterion/specification (e.g. „≥ 98.0%” or „Pass” with a definition),
- the result,
- the units,
- a reference to the method (SOP, pharmacopoeia, procedure number).
If a COA reports only the result without a specification, it is difficult to assess conformance. If only „Conforms/Pass” appears, this should derive from an explicit criterion rather than an assumption.
4) Analytical Methods on the COA (analytical methods listed on COA)
In the methods section, you should expect information such as:
- the technique (e.g. HPLC, LC–MS/MS, GC, NMR, ICP–MS),
- the method identifier (SOP / procedure number),
- the version or effective date (change control).
COA testing methods HPLC MS: what they typically mean
- HPLC is commonly used to assess the chromatographic profile and „chromatographic purity” (often reported as area% of peaks). It can indicate the presence of additional peaks but does not necessarily identify their nature.
- MS (mass spectrometry) is commonly used to assess whether the signal corresponds to the expected molecular mass. Depending on the method design, it may support identification but does not always exclude analogous compounds without adequate selectivity.
The term „HPLC” or „MS” alone is too generic to draw far-reaching conclusions without the context of the method.
5) Result Presentation: Units, Basis, and Rounding
Common sources of error:
- units (mg/mL vs mg/g, ppm vs µg/g),
- basis: „as is” vs „dry basis”,
- number of significant figures and rounding,
- qualifiers: „ND”, „<LOD”, „<LOQ”.
A result such as „99%” without clarifying whether it refers to assay (w/w) or area% by HPLC is ambiguous.
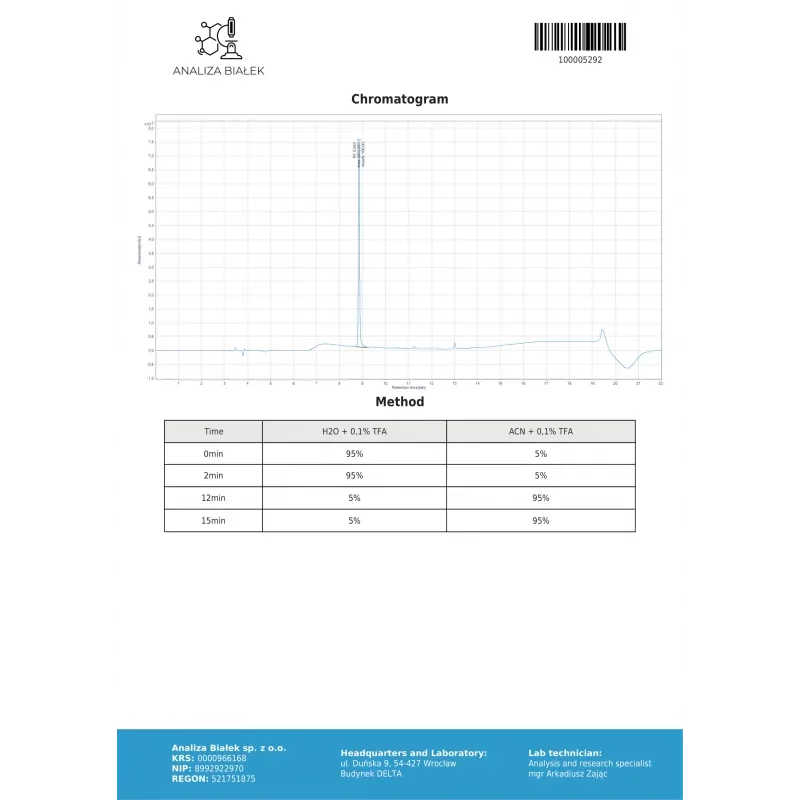
6) Supporting Data and Attachments (where available)
Some COAs additionally include:
- chromatograms,
- MS spectra,
- system suitability parameters (e.g. resolution, tailing),
- calibration information,
- identification of the reference standard.
The absence of attachments does not invalidate a COA, but it limits transparency and the ability to independently verify the context.
7) Document Authorisation and Integrity
Check that the COA has:
- a signature (electronic or handwritten) and the role of the authorising person,
- a report number and issue date,
- page numbering (e.g. „Page 1 of 2”),
- consistent formatting and the absence of contradictions.
Formal elements do not guarantee analytical correctness, but they are the foundation for assessing documentation quality.
How to Correctly Interpret COA Data (COA lab report explanation)
A COA should be read as a document describing a specific sample tested by a specific method at a specific point in time. This means conclusions should be limited to the scope of the report.
How to Interpret Identity Results (COA purity and identity)
An identity test typically answers the question: „Does the signal/analyte correspond to the expected material?”
- A „Pass” result may be based on retention time matching, spectral features, or a combination of criteria.
- Credibility increases when methods with different operating principles are used (e.g. separation + mass detection), but depends on selectivity and reference standards.
Limitation: identification is always „within the method.” The presence of a matching signal does not necessarily exclude all analogues or interferences if the method is not designed to address them.
How to Interpret „Purity” and „Assay” (certificate of analysis components)
In practice, different types of reporting are encountered:
- Assay (w/w or w/v): quantitative determination of analyte content using calibration.
- Chromatographic purity (area%): the proportion of the peak area relative to the total, dependent on separation conditions and detector response.
What can „99% purity” mean?
It may indicate a high proportion of the main peak under the given method conditions, but it does not necessarily imply:
- identification of all impurities,
- assessment of inorganic impurities,
- assessment of residual solvents,
- assessment of microbiological parameters,
- stability over time.
What Do ND, <LOD, and <LOQ Mean?
- ND (Not Detected): nothing observed above the detection threshold; does not mean „zero.”
- <LOD: below the limit of detection.
- <LOQ: below the limit of quantitation (may be detected but unreliable for quantitative reporting).
These are method- and sensitivity-dependent statements, not absolute assurances.
How to Read Accreditation and Scope Information
If the COA contains a reference to accreditation (e.g. ISO/IEC 17025), compliance interpretation should take into account that:
- accreditation typically covers a specific scope of methods, not everything the laboratory performs,
- the COA should allow assessment of whether the specific tests were within the accredited scope.
Most Common Mistakes in Interpreting a COA
Mistake 1: Treating a COA as a Guarantee of „Full Quality” or „Fitness for Purpose”
A COA documents the results of determinations within a defined scope. It is not a universal confirmation of all possible parameters.
Mistake 2: Assuming „Purity” Covers All Types of Impurities
„Purity” in a COA may refer only to the chromatographic profile. Other classes of impurities require separate methods.
Mistake 3: Overlooking Sampling and Representativeness
The result applies to the test sample; without information on the sampling plan and batch homogeneity, results should not automatically be generalised to the entire batch.
Mistake 4: Failing to Verify the Batch Number and Document Version
A lot/batch mismatch is the most common administrative irregularity. It must be ruled out before interpreting results.
Mistake 5: Misreading Units and the Basis of the Result
The most typical errors are confusing mg/mL with mg/g and assay with area%. In compliance contexts, ambiguities should not be interpreted in the most favourable light without clarification.
Mistake 6: Over-interpreting a Single Technique
MS can support identification but does not always exclude analogues; HPLC may show an absence of additional peaks under given conditions but does not guarantee detection of everything.
Limitations and Risks: What a COA May Not Cover
Even a correct COA may have limitations arising from:
- method scope (only selected parameters are analysed),
- sensitivity (LOD/LOQ),
- measurement uncertainty and variability,
- stability (the result is „as of the testing date”),
- level of detail (the certificate summarises data; raw data may be archived separately).
The compliance approach is to treat a COA as a document that helps assess conformance within the boundaries of the report, without extending conclusions beyond its scope.
Practical Conclusions for Informed Decisions
In practice, a COA is useful if it allows you to answer five control questions:
- Is the document assigned to the correct batch?
(batch/lot, sample ID, dates, report number) - Do the tests address the quality questions you want to answer?
(identity, assay/purity, specific impurities, etc.) - Are the acceptance criteria explicit, and does the result meet them?
(specification + result + units) - Are the methods and reporting approach sufficiently described?
(SOP, technique, version, LOD/LOQ, qualifiers) - Do you understand the limitations and avoid drawing conclusions beyond the COA’s scope?
(representativeness, analyte scope, stability)
If the answer to any of the above questions is negative, the correct stance is to treat the COA as insufficiently informative for strong conclusions.
Glossary of Terms Found on a COA
- COA / CoA: Certificate of Analysis.
- Batch/Lot number: batch identifier, production series identifier.
- Assay: quantitative determination of analyte content (typically with calibration).
- Area%: peak area proportion in chromatography (a relative measure within the method).
- HPLC: high-performance liquid chromatography.
- MS: mass spectrometry (detection based on m/z).
- LOD/LOQ: limit of detection / limit of quantitation.
- System suitability: criteria for correct performance of the analytical system.
Sources
ICH Q2(R1) – Validation of Analytical Procedures: Text and Methodology (principles of analytical method validation, including LOD/LOQ, specificity and „fitness for intended purpose”). database.ich.org
FDA – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients (section 11.4 „Certificates of Analysis” describes requirements for COA authenticity and information such as batch/lot). U.S. Food and Drug Administration
ISO/IEC 17025 – Testing and calibration laboratories (the role of the standard in demonstrating laboratory competence and result reliability). iso.org
European Commission – EudraLex Volume 4 (GMP guidelines) (reference point for the EU GMP approach, including parts covering active substances and quality documentation). Public Health
FDA – Bioanalytical Method Validation (Guidance for Industry) (indicates, among other things, the practice of having COA/purity data, stability, batch number, and manufacturer information for critical materials in studies). U.S. Food and Drug Administration